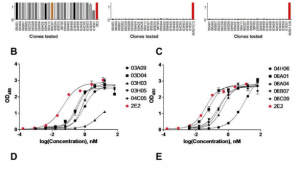
识别样品中细胞类型位置的新计算方法
斯坦福大学的研究人员开发了一种计算方法,用于在捕获空间转录组学时识别细胞在样本中的位置。该方法结合了来自空间转录的数据和参考单细胞RNA图谱,以创建建模输出。所得模型可用于查看细胞亚结构,识别共定位模式并按位置分析细胞类型内的差异表达。
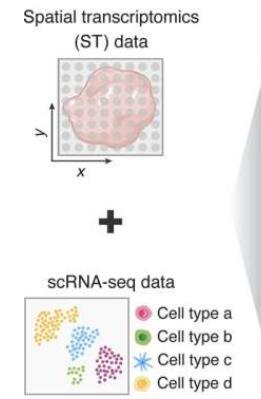
新方法 - 通过约束表达比对(CytoSPACE)的细胞空间定位分析 - 已发表在Nature Biotechnology杂志上。运行计算的脚本已通过GitHub免费提供。
医学研究对了解细胞内部和细胞之间的相互作用非常感兴趣。虽然基因组教会了我们很多关于遗传与疾病的联系,但基因在不同组织中的表达差异很大。局部细胞组织在预防或表现疾病中的作用对于理解、预测、诊断和治疗各种疾病至关重要。
组织内的细胞相互通信,发送群体或单个化学信息,微观管理细胞功能的健康运作。在癌症的情况下,通常是单个细胞内部的通信中断,这可能导致疾病形成,因为肿瘤细胞失去了听从群体停止生长和自我毁灭的指令的能力。
单细胞RNA测序可以捕获单个细胞中的基因表达(RNA分子),分辨率很高,可以与其他细胞进行比较。该技术从从感兴趣的组织中分离单个细胞开始。虽然它告诉研究人员有关测序的单个细胞内表达基因的所有信息,但它没有说明周围的细胞。
当前的空间转录组学方法对基因表达进行了更多的地理概述,以创建细胞关系图谱。研究人员获得的不是单个细胞信息,而是十个或更多有助于某个位置的细胞的样本。本质上,将组织样品放在将条形码附着在RNA上的RNA捕获载玻片上。当RNA从载玻片中取出并测序时,条形码可以追溯到其在载玻片上的收集位置,因此测序读数的空间排列可以重建为地图。
在寻找更准确和精确的数据时,已经开发了几种计算方法来推断空间转录组样品中的一般细胞组成。CytoSPACE利用来自参考scRNA-seq图谱的单细胞数据,并将其定位为更高的分辨率。
CytoSPACE构建了一组假定的输入单细胞序列,与空间转录组学样本中预测存在的内容相匹配。然后,它根据每个空间读取的预测细胞密度将这些序列填充到一组生成的位置。通过这些匹配的集合,CytoSPACE将组织重建任务作为线性分配问题,并以最佳方式将scRNA-seq数据中的单个细胞映射到空间坐标。
开发CytoSPACE的研究小组目前的研究针对几种领先的计算方法对其进行了测试。根据该论文,“在不同的平台和组织类型中,我们表明CytoSPACE在噪声耐受性和准确性方面优于以前的方法,能够以单细胞分辨率进行组织制图。
免责声明:本文为转载,非本网原创内容,不代表本网观点。其原创性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容、文字的真实性、完整性、及时性本站不作任何保证或承诺,请读者仅作参考,并请自行核实相关内容。